Pour vaincre les différents types de cancers du sang, les approches thérapeutiques de chimiothérapie sont largement utilisées en clinique. Toutefois, en raison de résistance aux traitements, le risque de rechute avec ces traitements est élevé. Les recherches actuelles tentent donc de découvrir des méthodes plus personnalisées et plus efficaces, notamment par l’entremise de certaines molécules capables de moduler la configuration spatiale de l’ADN. Ce pan de l’épigénétique, jusqu’ici très peu exploré, pourrait s’avérer prometteur dans la recherche sur la leucémie.
Depuis quelques semaines, Jérôme ressent une forte fatigue et cumule les infections. Ce sportif de 30 ans en bonne santé décide de consulter sa médecin de famille, qui lui fait passer une batterie de tests sanguins. Une prise de sang révèle un nombre anormalement faible de globules rouges et blancs qui explique la faiblesse généralisée et la succession des infections. La médecin soupçonne un cancer du sang. Une biopsie de la moelle osseuse confirme le diagnostic : Jérôme est atteint d’une leucémie myéloïde aiguë (LMA), qui représente 25 % de toutes les leucémies chez l’adulte[1]. La médecin juge que le jeune âge et l’état de santé de Jérôme favorisent ses chances de rémission. Pourtant, elle décide de réaliser des analyses génétiques afin de déceler des anomalies dans l’ADN de Jérôme, ce qui permettrait d’affiner la prise en charge thérapeutique. En se basant sur ces résultats, son équipe traitante pourrait proposer à Jérôme des traitements novateurs en cours d’essais cliniques.
Déchiffrer l’ADN
Le cancer du sang, ou leucémie, se caractérise par une multiplication incontrôlée des cellules souches * sanguines et un arrêt dans leur processus de maturation. Les cellules ainsi créées présentent une morphologie aberrante et remplacent progressivement les cellules sanguines fonctionnelles, une caractéristique facilement observable lors de la biopsie de la moelle osseuse de Jérôme.
Le traitement classique qui est proposé à Jérôme se base sur de la chimiothérapie cytotoxique *, qui consiste à éradiquer les cellules cancéreuses, et s’accompagne d’une greffe de cellules souches sanguines. Cependant, les résultats actuels de ces approches démontrent un manque d’efficacité en clinique. D’un cas clinique à l’autre, le pourcentage de rémission est très variable selon l’âge et le bagage génétique de la personne atteinte de leucémie, ce qui souligne le besoin de développer des solutions alternatives[2].
Mieux comprendre les mécanismes génétiques qui sous-tendent le développement de la LMA faciliterait la découverte d’approches thérapeutiques innovantes, comme le développement de nouvelles molécules pharmacologiques. Le séquençage * de l’ADN dans des cohortes de patients et de patientes leucémiques a mis en lumière la complexité génétique à l’origine de la maladie[3]. Cette analyse à grande échelle, menée en 2016 par plusieurs groupes de recherche à travers le monde, a révélé l’existence de sous-types de LMA, ce qui a amené la communauté scientifique à rechercher des traitements personnalisés. Selon la présence et l’accumulation de certaines mutations de gènes *, une classification permet maintenant d’estimer en clinique le pronostic vital, qui peut être plus ou moins favorable[4].
Dans le cas de Jérôme, comme pour environ une personne atteinte d’une LMA sur deux[5], le séquençage de son ADN a révélé que plusieurs mutations frappent un groupe de gènes ayant comme fonction d’assurer la lecture globale de l’information génétique. Les perturbations de ce mécanisme dans le développement d’une leucémie ne sont pas totalement comprises, mais les recherches à leur sujet pourraient conduire à de nouvelles solutions thérapeutiques en clinique.
Cibler le code histone
Une cellule humaine contient, à l’intérieur de son noyau, l’information génétique qui dictera l’intégralité de ses fonctions au sein d’un organe. Une cellule est composée de la molécule d’ADN, sur laquelle se trouvent environ 25 000 gènes qui peuvent être activés ou inactivés selon l’état physique et temporel de la cellule. Ainsi, les cellules de l’estomac, du cerveau ou du sang, par exemple, possèdent toutes la même molécule d’ADN avec un nombre identique de gènes. Chacun des gènes dicte le protocole qui mène à la production des protéines (comme des enzymes), qui sont les ouvrières des cellules. C’est la façon dont l’information génétique est lue par la cellule ainsi que la combinaison de gènes activés et inactivés qui dicteront la fonction spécifique de chacune des cellules.
Tel un réseau électrique, l’activation ou l’inactivation d’un gène est dépendante d’un système d’interrupteurs situé en amont de celui-ci. Chaque interrupteur doit être accessible physiquement pour qu’un ensemble de protéines, appelées « facteurs de transcription », puisse s’y fixer et l’actionner. Sans la présence des facteurs de transcription, l’activation génique est impossible. Or, la molécule d’ADN est compactée dans le noyau sous la forme d’une pelote de laine, nommée « chromatine ». Celle-ci est décrite comme un amas d’information génétique rassemblé grâce à des protéines, les « histones[6] ». Pour que les interrupteurs des gènes soient accessibles physiquement et que leur actionnement soit possible, la chromatine doit modifier sa conformation (ou organisation) et se dérouler. Ainsi, un gène activé et un gène réprimé se retrouvent respectivement dans une région de la chromatine déroulée ou enroulée (voir la figure 1). Ce processus qui agit sur les réarrangements spatiaux de la chromatine pour moduler les interrupteurs des gènes est l’un des volets d’étude de l’épigénétique *.
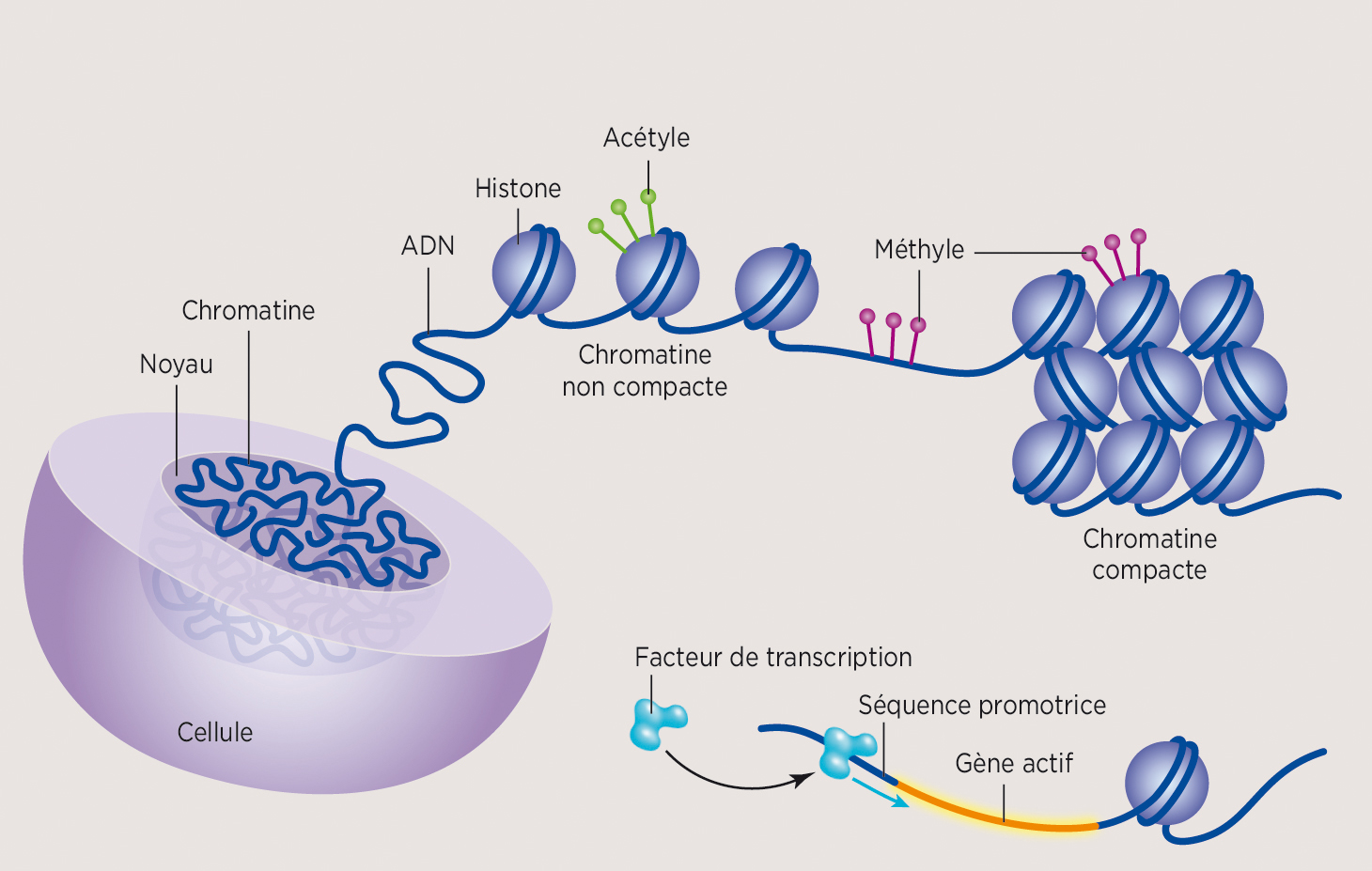
Illustration : Sylvie Dessert
Les recherches ont aussi montré qu’à la surface de la chromatine se trouvent des drapeaux de différentes couleurs qui forment un code histone[7]. Les drapeaux peuvent être ajoutés ou retirés par des enzymes, ce qui entraîne la reconfiguration spatiale de la chromatine, et ils indiquent si l’interrupteur du gène est accessible ou non. Ce code de couleurs permet ainsi d’associer ces marques d’histones à l’état d’enroulement de la pelote d’ADN et d’activité des gènes.
En s’appuyant sur ces diverses connaissances, la communauté scientifique qui s’intéresse aux leucémies tente de cibler le code histone et d’explorer de nouvelles stratégies thérapeutiques avec l’objectif de rétablir les positions et les présences de ces drapeaux, perturbées dans un contexte leucémique. Dans le cas de Jérôme, l’analyse menée par sa médecin a en effet mis en évidence une activation anormale de certains gènes ayant comme rôle de produire des enzymes responsables de la répartition de diverses marques d’histones. La conséquence directe de cette dérégulation est la reconfiguration de certaines régions de la chromatine. Cette reconfiguration entraîne alors l’activation anormale – en cascade – d’autres gènes, notamment ceux impliqués dans la maturation des cellules sanguines, ce qui est à l’origine d’un développement leucémique.
Bloquer les enzymes
La découverte de la dérégulation des acteurs épigénétiques dans les leucémies permet d’entrevoir un large éventail de perspectives thérapeutiques. Selon plusieurs études, cibler spécifiquement ces enzymes permettrait de restaurer le programme épigénétique ainsi que le niveau d’activation des gènes[8].
La thérapie épigénétique en est encore à ses débuts, mais le domaine est très prometteur et en constante évolution. De nombreuses cibles thérapeutiques épigénétiques sont en cours d’évaluation pour traiter les LMA. C’est le cas de la famille des histones déméthylases *, composée notamment de la protéine KMD1A, connue pour jouer un rôle essentiel dans la production des cellules sanguines et le retrait de groupements méthyle des histones. En clinique, certains groupes de patientes et de patients leucémiques présentent une activité anormale de KDM1A qui entraîne une accumulation de cellules immatures et bloque le processus de maturation des cellules[9]. Développer un inhibiteur ciblant spécifiquement KMD1A semble donc une avenue thérapeutique prometteuse : des essais cliniques sont en cours d’évaluation en Europe alors que les premiers résultats des données précliniques amènent une lueur d’espoir[10].
D’autres études présentent des résultats plus mitigés, entre autres celles concernant la protéine DOT1L, une histone méthyltransférase * capable de rajouter un groupement méthyle sur certaines histones de la chromatine et ainsi assurer une activation génique. En clinique, des données démontrent que l’activation anormale de DOT1L entraîne des niveaux élevés de marques d’histones qui sont responsables d’une hausse aberrante de l’activation de gènes cibles[11]. La molécule pharmacologique étudiée, nommée « pinométostat », agit comme un inhibiteur de l’enzyme[12], mais l’issue des études cliniques reste encore incertaine. Bien qu’une baisse de la présence des groupements méthyle sur la chromatine soit observée dans ces études, l’absence de réponses cliniques dans la cohorte des personnes y ayant participé laisse à penser que des études complémentaires sont nécessaires pour confirmer la pertinence de choisir cette cible en clinique[13]. Cet exemple démontre la complexité de trouver une cible thérapeutique intéressante et le besoin d’aller explorer de nouvelles pistes de solution.
Depuis 2017, de nouveaux médicaments inhibiteurs ont été autorisés par la Food and Drug Administration, aux États-Unis. L’un d’entre eux cible spécifiquement les gènes IDH1 et IDH2. Bloquer l’action du produit de ces gènes est un exemple de thérapie épigénétique porteur d’espoir puisque les études démontrent une réponse favorable en clinique et une augmentation des taux de survie chez les malades. Par un mécanisme d’action touchant le métabolisme des cellules, ces inhibiteurs sont capables de corriger les programmes d’activation génique altérés par des changements dans les marques d’histones, mais également dans la méthylation de l’ADN, un autre versant de l’épigénétique sur lequel la communauté scientifique s’attarde plus particulièrement[14].
Combiner les stratégies
Une autre approche consisterait à combiner les traitements déjà existants en clinique avec les thérapies ciblant les mécanismes épigénétiques. Évaluer l’activité d’un agent thérapeutique seul est certes une première étape essentielle, mais tester son efficacité en combinaison avec d’autres stratégies, comme l’immunothérapie *, permettrait de déterminer le portrait optimal du traitement. À titre d’exemple, la molécule inhibitrice de DOT1L est actuellement en essai clinique en association avec une chimiothérapie conventionnelle dans un type de LMA.
Malgré des progrès importants dans la compréhension des mécanismes génétiques de la leucémie myéloïde aiguë, les options thérapeutiques restent toujours limitées. Les traitements par chimiothérapie se heurtent à l’enjeu des effets secondaires et du développement des résistances aux médicaments. La leucémie étant une maladie complexe, la médecine personnalisée viserait à choisir judicieusement un traitement en fonction du profil génétique de chaque personne atteinte. Les équipes traitantes peuvent difficilement prédire les événements de rechute, qui demanderaient chaque fois une nouvelle analyse génétique pour qu’elles puissent choisir un traitement adapté. Toutefois, les scientifiques persistent et tentent d’optimiser les stratégies thérapeutiques, entre autres en démêlant les gènes pour mieux combattre les leucémies.
Lexique :
* Cellule souche : cellule immature capable de se spécialiser et de générer les différents types de cellules du système sanguin (globules rouges, globules blancs, plaquettes, etc.).
* Chimiothérapie cytotoxique : traitement clinique visant à empêcher les cellules de se diviser. L’objectif est de détruire les cellules cancéreuses, mais la stratégie n’est pas sans conséquence sur les cellules non cancéreuses.
* Séquençage : détermination de la séquence de l’ADN grâce à l’utilisation d’un outil appelé « séquenceur » et d’outils bioinformatiques.
* Mutation de gène : modification de la molécule d’ADN qui pourrait avoir comme conséquence le développement d’un cancer.
* Épigénétique : étude des changements réversibles dans l’activité de gènes qui n’impliquent pas de modification dans la séquence d’ADN en elle-même. L’épigénétique peut s’intéresser, par exemple, aux modifications des histones, à la méthylation de l’ADN et aux petits ARN non codants.
* Déméthylase : enzyme qui assure le retrait du groupement méthyle.
* Méthyltransférase : enzyme qui assure l’ajout du groupement méthyle.
* Immunothérapie : traitement visant à stimuler l’immunité de l’organisme par l’administration d’anticorps ou d’antigènes.
Références
[1] O’Donnell, M. R., Abboud, C. N., Altman, J., Appelbaum, F. R., Arber, D. A., Attar, E., Borate, U., Coutre, S. E., Damon, L. E., Goorha, S., Lancet, J., Maness, L. J., Marcucci, G., Millenson, M. M., Moore, J. O., Ravandi, F., Shami, P. J., Smith, B. D., Stone, R. M., Strickland, S. A., … Gregory, K. M. (2012). NCCN Clinical Practice Guidelines Acute myeloid leukemia. Journal of the National Comprehensive Cancer Network: JNCCN, 10(8), 984-1021.
[2] Lokody, I. (2014). Drug resistance: Overcoming resistance in acute myeloid leukaemia treatment. Nature Reviews Cancer, 14(7), 452-453.
Shah, A., Andersson, T. M., Rachet, B., Bjorkholm, M. et Lambert, P. C. (2013). Survival and cure of acute myeloid leukaemia in England, 1971-2006: A population-based study. British Journal of Haematology, 162(4), 509-516.
[3] Papaemmanuil, E., Dohner, H. et Campbell, P. J. (2016). Genomic classification in acute myeloid leukemia. The New England Journal of Medicine, 375(9), 900-901.
[4] Döhner, H., Estey, E. H., Amadori, S., Appelbaum, F. R., Büchner, T., Burnett, A. K., Dombret, H., Fenaux, P., Grimwade, D., Larson, R. A., Lo-Coco, F., Naoe, T., Niederwieser, D., Ossenkoppele, G. J., Sanz, M. A., Sierra, J., Tallman, M. S., Löwenberg, B. et Bloomfield, C. D. (2010). Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood, 115(3), 453-474.
[5] Papaemmanuil et al. (2016), op. cit.
[6] Dawson, M. A., Kouzarides, T. et Huntly, B. J. (2012). Targeting epigenetic readers in cancer. The New England Journal of Medicine, 367(7), 647-657.
[7] Jenuwein, T. et Allis, C. D. (2001). Translating the histone code. Science, 293(5532), 1074-1080.
[8] Cai, S. F., Chen, C. W. et Armstrong, S. A. (2015). Drugging chromatin in cancer: Recent advances and novel approaches. Molecular Cell, 60(4), 561-570.
[9] Harris, W. J., Huang, X., Lynch, J. T., Spencer, G. J., Hitchin, J. R., Li, Y., Ciceri, F., Blaser, J. G., Greystoke, B. F., Jordan, A. M., Miller, C. J., Ogilvie, D. J. et Somervaille, T. C. (2012). The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell, 21(4), 473-487.
[10] Maes, T., Mascaro, C., Tirapu, I., Estiarte, A., Ciceri, F., Lunardi, S., Guibourt, N., Perdones, A., Lufino, M., Somervaille, T., Wiseman, D. H., Duy, C., Melnick, A., Willekens, C., Ortega, A., Martinell, M., Valls, N., Kurz, G., Fyfe, M., Castro-Palomino, J. C., … Buesa, C. (2018). ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell, 33(3), 495-511 e412.
[11] Okada, Y., Feng, Q., Lin, Y., Jiang, Q., Li, Y., Coffield, V. M., Su, L., Xu, G. et Zhang, Y. (2005). hDOT1L links histone methylation to leukemogenesis. Cell, 121(2), 167-178.
[12] Stein, E. M., Garcia-Manero, G., Rizzieri, D. A., Tibes, R., Berdeja, J. G., Savona, M. R., Jongen-Lavrenic, M., Altman, J. K., Thomson, B., Blakemore, S. J., Daigle, S. R., Waters, N. J., Suttle, A. B., Clawson, A., Pollock, R., Krivtsov, A., Armstrong, S. A., DiMartino, J., Hedrick, E., Löwenberg, B., … Tallman, M. S. (2018). The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood, 131(24), 2661-2669.
[13] Ibid.
[14] Stein, E. M., DiNardo, C. D., Pollyea, D. A., Fathi, A. T., Roboz, G. J., Altman, J. K., Stone, R. M., DeAngelo, D. J., Levine, R. L., Flinn, I. W., Kantarjian, H. M., Collins, R., Patel, M. R., Frankel, A. E., Stein, A., Sekeres, M. A., Swords, R. T., Medeiros, B. C., Willekens, C., Vyas, P., … Tallman, M. S. (2017). Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood, 130(6), 722-731.